
https://doi.org/10.1140/epjp/i2018-12036-8
Regular Article
First-principles study of In mXn (X = Se, Te; m + n = 5 clusters: Structural, electronic, magnetic and spectral properties
1
Department of Applied Physics, School of Physics, University of Electronic Science and Technology of China, 610054, Chengdu, China
2
School of Sciences and Research Center for Advanced Computation, Xihua University, 610039, Chengdu, China
* e-mail: zhongsiying68@163.com
Received:
9
February
2018
Accepted:
26
April
2018
Published online:
1
June
2018
Using the density functional theory (DFT) method, the geometrical structures of 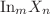 (
(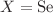 , Te;
, Te; 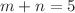 clusters are optimized, and their relative stability as well as electronic, magnetic and spectral properties are calculated. The ground state structures of clusters are found to be largely similar for and Te, with the exception of
clusters are optimized, and their relative stability as well as electronic, magnetic and spectral properties are calculated. The ground state structures of clusters are found to be largely similar for and Te, with the exception of 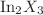 . The energy gap exhibits the maximum for
. The energy gap exhibits the maximum for 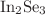 or
or 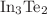 . The electronic properties of clusters depend on their geometrical structures and, hence, on the value of m ; and shows a low vertical electron affinity (VEA) of about 1.60eV and a high vertical ionization potential (VIP) of about 9.33eV. The total magnetic moment is 1 or 0μ_B for the clusters with
. The electronic properties of clusters depend on their geometrical structures and, hence, on the value of m ; and shows a low vertical electron affinity (VEA) of about 1.60eV and a high vertical ionization potential (VIP) of about 9.33eV. The total magnetic moment is 1 or 0μ_B for the clusters with 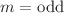 (1, 3) or even (2, 4) , respectively. The local magnetic moments of X atoms amount to about 99.9% of the total magnetic moment, while those of In atoms are merely 0.1%. The IR and Raman spectra of clusters exhibit similarity for and Te with an exception of . The energies of the strongest peaks of
(1, 3) or even (2, 4) , respectively. The local magnetic moments of X atoms amount to about 99.9% of the total magnetic moment, while those of In atoms are merely 0.1%. The IR and Raman spectra of clusters exhibit similarity for and Te with an exception of . The energies of the strongest peaks of 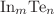 are largely smaller than the corresponding
are largely smaller than the corresponding 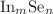 in both IR and Raman spectra. For UV-Vis spectra, the absorption peaks at 200-400nm for all clusters and 390–780 nm for m = 1 and 3 (except are likely to hint useful properties of UV and visible light absorption, respectively.
in both IR and Raman spectra. For UV-Vis spectra, the absorption peaks at 200-400nm for all clusters and 390–780 nm for m = 1 and 3 (except are likely to hint useful properties of UV and visible light absorption, respectively.
© Società Italiana di Fisica and Springer-Verlag GmbH Germany, part of Springer Nature, 2018


