
https://doi.org/10.1140/epjp/i2017-11733-0
Regular Article
Structural and electronic properties of CuxAg1-xCl: First-principles study
1
Laboratoire de Micro et de Nanophysique LaMiN - ENP d’ORAN, BP 1523, 31000, El M’Naouer, Algeria
2
Faculté de SNVST, Université AKLI Mohand-Oulhadj, Bouira, Algeria
3
Laboratoire de Modélisation et de Simulation en Sciences des Matériaux, Université Djillali Liabès, Sidi Bel-Abbes, Algeria
4
LPMF USTO-MB BP 1505 Oran El M’Naouer, 31000, Oran, Algeria
* e-mail: fasbenplast@yahoo.fr
Received:
22
March
2016
Accepted:
2
October
2017
Published online:
9
November
2017
The structural and electronic properties of the ternary 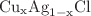 alloy are investigated using a recent version of the full potential linear muffin-tin orbitals method (FPLMTO) based on the density functional theory, within both the local density approximation (LDA) and the generalized gradient approximation (GGA). The lattice constants, bulk modulus and band gap were calculated as a function of copper molar fraction x in rock salt (B1) and zincblende (B3) structures. These parameters were found to depend non-linearly on alloy concentration Cu, except for the lattice parameter which follows Vegard’s law. Our results predict the rock salt phase as the ground state for this ternary system.
alloy are investigated using a recent version of the full potential linear muffin-tin orbitals method (FPLMTO) based on the density functional theory, within both the local density approximation (LDA) and the generalized gradient approximation (GGA). The lattice constants, bulk modulus and band gap were calculated as a function of copper molar fraction x in rock salt (B1) and zincblende (B3) structures. These parameters were found to depend non-linearly on alloy concentration Cu, except for the lattice parameter which follows Vegard’s law. Our results predict the rock salt phase as the ground state for this ternary system.
© Società Italiana di Fisica and Springer-Verlag GmbH Germany, part of Springer Nature, 2017


