
https://doi.org/10.1140/epjp/i2016-16285-1
Regular Article
Geometrical structure, stability and electronic properties of AunHg( ) clusters
) clusters
1
School of Science, Southwest University of Science and Technology, 621010, Mianyang, Sichuan, China
2
College of Physics, Chongqing University, 400044, Chongqing, China
* e-mail: wanwei@swust.edu.cn
Received:
30
January
2016
Accepted:
18
July
2016
Published online:
24
August
2016
The geometrical structures, relative stabilities, electronic properties and chemical hardness of AunHg(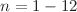 ) clusters are systematically investigated using the density functional theory with relativistic all-electron methods. The optimized low-lying energy geometries exhibit two-dimensional and three-dimensional structures. Furthermore, all the lowest-energy structures of AunHg() clusters favor planar geometries with slight distortion, in which the dopant Hg atom prefers to occupy a peripheral site with a lower coordination. The geometrical, electronic and chemical stabilities of the AunHg cluster with even number of valence electrons are higher than those of the neighboring AunHg cluster with odd number of valence electrons. Besides, 5d valence electrons of impurity Hg atom in the AunHg cluster hardly join in the orbital interactions compared with 5d valence electrons of corresponding Au atom in Aun+1 cluster. Au-Hg bonds in AunHg clusters are weaker and have more obviously ionic-like characteristics than the corresponding Au-Au bonds in Aun+1 clusters.
) clusters are systematically investigated using the density functional theory with relativistic all-electron methods. The optimized low-lying energy geometries exhibit two-dimensional and three-dimensional structures. Furthermore, all the lowest-energy structures of AunHg() clusters favor planar geometries with slight distortion, in which the dopant Hg atom prefers to occupy a peripheral site with a lower coordination. The geometrical, electronic and chemical stabilities of the AunHg cluster with even number of valence electrons are higher than those of the neighboring AunHg cluster with odd number of valence electrons. Besides, 5d valence electrons of impurity Hg atom in the AunHg cluster hardly join in the orbital interactions compared with 5d valence electrons of corresponding Au atom in Aun+1 cluster. Au-Hg bonds in AunHg clusters are weaker and have more obviously ionic-like characteristics than the corresponding Au-Au bonds in Aun+1 clusters.
© Società Italiana di Fisica and Springer-Verlag Berlin Heidelberg, 2016


